ShotGun and DesignPlanner Tutorial
What You Need
Installation
-
The current beta release of ShotGun is static executable and is ready to use (no need for installation).
You will only need to extract the files by using the following command line:
tar -zxvf ShotGun.*.tgz
How to Run
ShotGunOnly ShotGun+DesignPlannerShotGun Only
Options Command DocumentationAvailable Options:

Command Line Example:
Under csh:set d = ./2012-03-29_ShotGun
set hf = $d/data/canHap/chrs44880.1.canHap.gz
set pf = $d/data/simp_pos/EU.1.pos
$d/bin/ShotGun -h $hf -p $pf -n 60 -d 6 -o n60.depth6
Under bash:
d=./2012-03-29_ShotGun
hf=$d/data/canHap/chrs44880.1.canHap.gz
pf=$d/data/simp_pos/EU.1.pos
$d/bin/ShotGun -h $hf -p $pf -n 60 -d 6 -o n60.depth6
The command line above generates average 6X sequencing data for 60 individuals.
Full Documentation for the options:
- --hapfile[-h] : Required input haplotype file
example w/ 20 chromosomes at 50 markers - --posfile[-p] : Required input position file
example for 50 markers - --errorfile : Optional input cycle-specific error file
example for 35-base reads - --seed : Random seed, integer, default = 12345
- --readlength[-l] : Read Length, positive integer, default = 100
- --minwts : Recommend using --alpha (below) instead.
- --BeginningPosition[b] : Start Position, positive number, default = 1.0
- --EndingPosition[e] : End Position, positive number, default = 100000.0
- --sequencableProportion: Sequencable Proportion, default = 1.0
- --alpha[a] : False Positive Rate, default = 1e-3
1e-3 corresponds to 100 false positives in a 100Kb region - --shape[s] : Shape Parameter for Gamma Mixture.
Default = 4.0, also see --negativeBinomial.
Smaller value => more severe over-dispersion compared with Poisson. - --numIndividuals[n] : Number of individuals to be sequenced
positive integer, default = 100 - --depth[d] : Average Sequencing Depth per Individual.
- --erate : Average Sequencing Error Rate, ignored if --errorfile provided.
- --allbps : Output Information for ALL Base Pairs,
regardless of evidence for being polomorphic. - --negativeBinomial : Depths follow Negative Binomial distribution.
Implemented as a Gamma mixture of Poisson's.
Shape parameter of Gamma specified by --shape. - --trueped : Output true genotype information at detected markers.
- --numReplications : #Replicates to determine polymorphism score threshold.
- --prefix : Output prefix.
ShotGun+DesignPlanner
CommandLine Options Output Interpretation OutputFieldsCommand Line Example:
Under csh:set d = ./2012-03-29_ShotGun
cd $d
./DesignPlanner.csh 0
Under bash:
d=./2012-03-29_ShotGun
cd $d
./DesignPlanner.csh 0
The command line above will generate effective sample size estimate for each MAF category for the default setting: 30 individuals sequenced at depth 5X (you can change the parameters by changing the 26th and 28th line in the shell script), with a ShotGun false positve rate of ~1/1,000 (that is, ~100 false positive SNPs in each 100Kb region).
Note that the 0 at the end of the command line is for setting warning option. By default warning = 1, which means the program will STOP for warning.
See example screen output after running a similar command line (60 individuals at depth 2X).
Options:
Options are specified in DesignPlanner.csh. You will need to modify the codes inside the shell script, in the "SET PARAMETERS" sections at the beginning of the shell script. The following options are available:- n : #individuals
- t : depth
- alpha: false positive rate (see ShotGunDocumentation)
- erate: Average Sequencing Error Rate (see ShotGunDocumentation)
- st : --states for thunder
larger is better at the cost for quadratically increased computing time - rd : --rounds for thunder
larger is better at the cost for linearly increased computing time
Example Output:
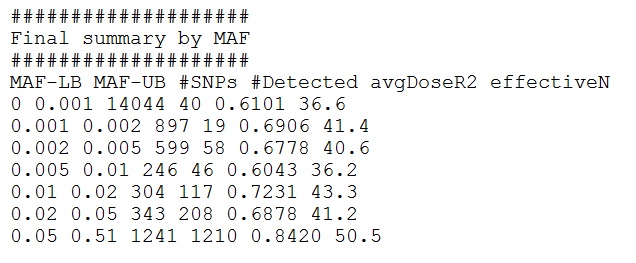
Example Output Interpretation:
-
Selected interpretation of the output: using the design of 60 individuals sequenced at an average depth of 2X, While only a small percentage of rare markers (e.g.,
19 / 897 = ~2% of markers with MAF 0.001-0.002) can be detected, almost all the common markers (e.g., 1210 / 1241 = ~97.5% of markers with MAF > 0.05) can be detected.
In addition, the quality of genotype calls (measured by information content dosage r2 or effective sample size) also tends to improve with increasing MAF. For
example, the effective sample size is only ~2/3 of the sequenced (41.4 compared with the 60 sequenced) for markers with MAF 0.001-0.002; and the effective sample size
is ~5/6 for markers with MAF > 0.05 (50.5 compared with 60).
Note that the pattern fluctuation is caused by chance due to small number of markers involved in some MAF categories.
Output Fields:
- MAF-LB : Minor Allele Frequency (MAF) Lower Bound
- MAF-UB : MAF Upper Bound
- #SNPs : Number of SNPs with "Population"a MAF in the Category
- #Detected : Number of SNPs Detected by the Designb
- avgDoseR2 : Average Dosagec r2d for the Detected SNPs
- effectiveN : Effective Sample Size = n * avgDoseR2
-
a: population = 45K chromosomes
b: by default, the design is 60 individuals sequenced at depth 2X
c: Dosage, ranging continuously from 0 to 2, is the fractional count of a designated allele.
d: Dosage r2, a marker level quality metric for information content, is the squared Pearson correlation between the dosages and true genotypes.